Wir navigieren Sie durch das Labyrinth der Vorschriften für Medizinprodukte
Willkommen in der Welt der Vorschriften für Medizinprodukte! Als Hersteller von Medizinprodukten oder als Student:in in der Branche haben Sie wahrscheinlich schon von der ISO 13485, der FDA-Norm 21 CFR Part 820 und der Europäischen Medizinprodukteverordnung (MDR, Medical Device Regulation) gehört. Diese Normen sind so etwas wie die Leitsterne für die Gewährleistung von Sicherheit, Qualität und Compliance. In diesem Blogbeitrag werden wir diese komplexen Vorschriften auf ansprechende und leicht verständliche Weise enträtseln. Los geht’s, tauchen wir tief in jede dieser wichtigen Normen ein!
Beginnen wir mal mit der ISO-Norm: Was ist ISO 13485 überhaupt? Stellen Sie sich ISO 13485 als die universelle Sprache des Qualitätsmanagements für Medizinprodukte vor. Es handelt sich um eine international anerkannte Norm, die die Anforderungen an ein robustes Qualitätsmanagementsystem (QMS) festlegt. Ganz gleich, ob Sie in Tokio, Toronto oder Timbuktu ansässig sind, die ISO 13485 trägt dazu bei, dass Ihr Medizinprodukt die Erwartungen der Kunden und der Behörden durchgängig erfüllt.

1. Qualitätsmanagementsystem (QMS): Betrachten Sie dies als das Rückgrat Ihres Betriebs. ISO 13485 bietet einen detaillierten Fahrplan für die Einrichtung eines QMS, das alle Bereiche vom Entwurf bis zur Auslieferung abdeckt.
2. Verantwortung des Managements: Die obersten Führungskräfte müssen für die Qualität einstehen. Sie geben den Ton an mit einer Qualitätspolitik, spezifischen Zielen und regelmäßigen Kontrollen, um sicherzustellen, dass alles auf Kurs ist.
3. Ressourcenmanagement: Einige wichtige Punkte der Regulatorien lassen sich am besten mit einem Vergleich erklären. Jede:r war doch schon mal in einem Restaurant. Die Abläufe dort sind einigermaßen vertraut. Stellen Sie sich das Ressourcenmanagement so vor wie die Ausstattung der Küche und die Sorge, dass die Köche geschult sind. Sie brauchen die richtigen Mitarbeiter:innen, die richtige Infrastruktur und das richtige Umfeld, um Qualitätsprodukte herzustellen.
4. Produktrealisierung: Dies ist das Rezept für Ihr Gerät – Planung, Design, Beschaffung, Fertigung und Überwachung. Jeder Schritt stellt sicher, dass Ihr Produkt genau richtig wird.
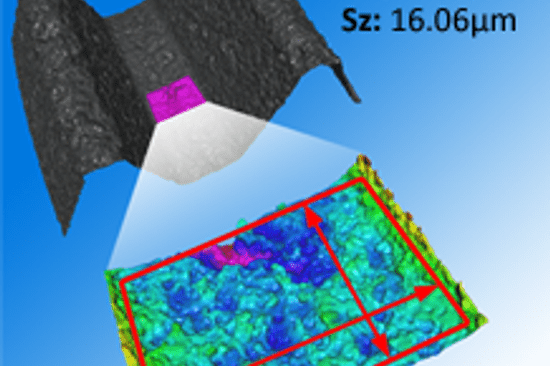
5. Messung, Analyse und Verbesserung: Ständige Geschmackstests und Optimierungen. Sie überwachen Prozesse und Produkte, führen Audits durch, kümmern sich um Nichtkonformitäten und verbessern sich kontinuierlich. Genau hier sind die Systeme von Bruker Alicona zu Hause. Gemessen werden aber nicht Geschmack und Geruch von medizinischen Produkten, sondern die Maß, Lage, Form und Rauheit. Warum gerade die Rauheit so entscheidend ist, lesen Sie in diesem Artikel.
Globale Anerkennung
Weltweite Bestätigungen, die den Eintritt in verschiedene Märkte erleichtern.
Vertrauen der Kund:innen:
Zeigt Ihr Engagement für Qualität und schafft Vertrauen bei Kunden und Aufsichtsbehörden.
Kontinuierliche Verbesserung
Fördert eine Kultur der ständigen Verbesserung.
Branchengültigkeit
Was die Zertifizierung angeht, sitzen alle im selben Boot, es gibt kein Entkommen und niemanden, der strengere oder weniger strenge Auflagen hat. Seien wir ehrlich: Ist es nicht besser, eine solche Tatsache cool zu finden als sich über die Anstrengungen aufzuregen?
In den USA müssen Produzenten von Medizinprodukten die FDA-Vorschrift 21 CFR Part 820, auch bekannt als Quality System Regulation (QSR), befolgen. Es handelt sich um eine umfassende Checkliste, mit der Sie garantieren können, dass Ihre Produkte sicher und wirksam sind, bevor sie auf den Markt kommen.
1. Anforderungen an das Qualitätssystem: Ähnlich wie bei ISO 13485 wird hier der gesamte Lebenszyklus eines Produkts abgedeckt, vom ersten Konzept bis zum Verkauf.
2. Konstruktionskontrollen: Stellt sicher, dass die Geräte so konzipiert sind, dass sie den Bedürfnissen der Benutzer und der beabsichtigten Verwendung entsprechen. Es ist wie beim Bau eines Hauses – detaillierte Pläne sind unerlässlich.
3. Dokumentenkontrollen: Ordnungsgemäße Dokumentation für alles. Bleiben wir doch direkt bei unserem Restaurant-Beispiel: Stellen Sie sich vor, Sie führen akribisch Buch über Ihre Rezepte und Prozesse.
4. Einkaufskontrollen: Stellt sicher, dass das, was Sie kaufen (in unserem Fall die Zutaten), den festgelegten Anforderungen entspricht.
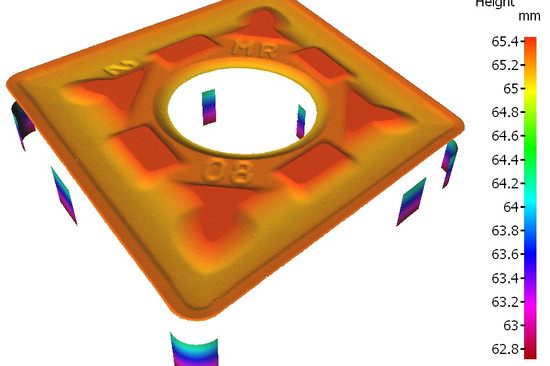
5. Produktions- und Prozesskontrollen: Validiert Prozesse, um eine gleichbleibende Qualität zu gewährleisten. Das ist wie ein Koch, der ein Gericht jedes Mal perfektioniert. Oder wie das Bruker Alicona Messsystem, das jede Abweichung von der Zeichnung sofort erkennt.
6. Korrektur- und Vorbeugungsmaßnahmen (CAPA): Untersucht Nichtkonformitäten, ermittelt die Grundursachen und führt Korrekturen durch. Zum Beispiel, um herauszufinden, warum ein Gericht misslungen ist, und um sicherzustellen, dass es nicht wieder vorkommt.
7. Aufzeichnungen und Berichte: Führt detaillierte Aufzeichnungen und berichtet über Probleme wie unerwünschte Ereignisse. Stellen Sie sich vor, Sie führen ein tägliches Tagebuch über Ihre kulinarischen Erlebnisse.
Im Jahr 2022 schlug die FDA vor, 21 CFR Part 820 mit der ISO 13485 zu harmonisieren, indem sie die Quality Management System Regulation (QMSR) einführte. Diese Änderung zielt darauf ab, die Anforderungen der FDA enger an die internationale Norm anzugleichen und die regulatorische Belastung für globale Hersteller zu verringern.
Angleichung an ISO 13485:
QMSR übernimmt das Rahmenwerk der ISO 13485 und vereinfacht so die Einhaltung der Vorschriften.
Vereinfachte Anforderungen:
Reduzierung der einzelnen FDA-Anforderungen und Konzentration auf den umfassenden Ansatz der ISO 13485.
Konsistenz:
Leichtere Verwaltung der globalen Konformität für Unternehmen.
Am 2. Februar 2024 hat die FDA die finale Überarbeitung der QSR (21 CFR Part 820) und der 21 CFR Part 4 (Combination products) in Form der Final Rule 89 FR 7496 im Federal Register mit Gültigkeit ab 2. Februar 2026 veröffentlicht.

Die Europäische Medizinprodukteverordnung (MDR) ist der strenge Rahmen der EU für Medizinprodukte. Sie ersetzt die alte Medizinprodukterichtlinie (MDD) und bedeutet, dass die EU die Anforderungen an Sicherheit und Leistung erhöht hat.
1. Geltungsbereich und Klassifizierung: Breiterer Geltungsbereich und Neueinstufung von mehr Produkten in höhere Risikokategorien. Das ist so, als würde man eine Kücheninspektion von einer Routinekontrolle in eine Superprüfung umwandeln.
2. Klinische Bewertung und Untersuchung: Erfordert solide klinische Daten zum Nachweis von Sicherheit und Leistung. Stellen Sie sich das wie ausführliche Geschmackstests und Bewertungen vor der Markteinführung eines Gerichts vor.
3. Post-Market Surveillance: Kontinuierliche Überwachung der auf dem Markt befindlichen Produkte. Es ist wie in einem Restaurant, das ständig das Feedback der Kunden prüft und Anpassungen vornimmt.
4. Einmalige Gerätekennzeichnung (UDI = Unique Device Identification): Ein System zur Rückverfolgung von Geräten. Stellen Sie sich vor, jede Zutat wird zur Rückführbarkeit mit einem Etikett versehen.
5. Notifizierte Stellen: Gestärkte Rolle bei der Konformitätsbewertung. Das ist so, als würden Meisterköche Ihre Küche und Rezepte bewerten.
6. Technische Dokumentation: Detaillierte Dokumentationsanforderungen. Stellen Sie sich vor, Sie stellen ein umfassendes Kochbuch zusammen, das jedes noch so kleine Detail enthält.
Verbesserte Sicherheit
Strengere Anforderungen sorgen für höhere Sicherheits- und Leistungsstandards.
Transparenz und Rückführbarkeit
Verbesserte Transparenz durch UDI und öffentlichen Zugang zu Sicherheitsdaten.
Globale Wettbewerbsfähigkeit
Angleichung an globale Normen, Erleichterung des internationalen Handels.

Schwerpunkt Qualitätsmanagement: Alle drei betonen die Notwendigkeit eines soliden QMS.
Lebenszyklus-Abdeckung: Sie decken den gesamten Lebenszyklus eines Produkts ab, von der Auslegung bis zu Aktivitäten nach dem Inverkehrbringen.
Risikomanagement: Gewährleistet, dass die Produkte sicher und wirksam sind, mit robusten Risikomanagementverfahren.
Geografischer Geltungsbereich: ISO 13485 ist global, 21 CFR Part 820 ist US-spezifisch, und die MDR gilt in der EU.
Regulierungsbehörden: Die FDA beaufsichtigt 21 CFR Part 820/QMSR, während die EU benannte Stellen für die MDR einsetzt.
Spezifische Anforderungen: Während ISO 13485 und 21 CFR Part 820/QMSR eng aneinander angeglichen sind, enthält die MDR einzigartige Elemente wie UDI und strengere klinische Bewertungen.
Der Schritt der FDA, 21 CFR Part 820 mit ISO 13485 durch QMSR zu harmonisieren, ist ein großer Gewinn für die Hersteller. Dies bedeutet eine übersichtlichere Regulierungslandschaft und weniger Redundanz, was die Einhaltung sowohl der amerikanischen als auch der internationalen Normen erleichtert. Auch die EU täte gut daran, die MDR mit dem ISO-Standard in Einklang zu bringen.
Für Hersteller von Medizinprodukten und solche, die es werden wollen, ist das Verständnis von ISO 13485, 21 CFR Part 820 und der MDR von entscheidender Bedeutung, um sich in dem komplexen Labyrinth der Vorschriften zurechtzufinden. Diese Normen und Vorschriften gewährleisten die Sicherheit, Qualität und Wirksamkeit von Medizinprodukten und dienen letztlich dem Schutz der Patienten und dem Erfolg der Unternehmen auf den globalen Märkten. Die Harmonisierung der FDA-Vorschriften mit der ISO 13485 ist ein bedeutender Schritt nach vorn, der die Einhaltung der Vorschriften vereinfacht und einen einheitlicheren Ansatz für das Qualitätsmanagement in der Medizinproduktebranche fördert. Unabhängig davon, ob Sie das nächste große Ding in der Medizintechnik entwickeln oder sich auf diesem dynamischen Gebiet weiterbilden, wird Ihnen die Kenntnis dieser Vorschriften dabei helfen, sicherzustellen, dass Ihre Produkte nicht nur innovativ, sondern auch sicher, effektiv und konform sind. Viel Spaß beim Forschen, Entwickeln und Produzieren!
dieSonne-web-(21)-550x366.jpg "Turbine Blade with Cooling Holes")




dieSonne-(10)-2076x1706-550x366.jpg)
-550x366.jpeg)
die_Sonne-550x366.png "Bruker Alicona at AMB 2024 in Stuttgart")



_Nagel-550x366.jpg)


-1024x576-550x366.jpg)
dieSonne-065-550x366.jpg)

dieSonne-120-550x366.jpg)






dieSonne-137-550x366.jpg)




dieSonne-web-(102)-550x366.jpg)
-550x366.jpg)
dieSonne-(23)-1706x1708-550x366.jpg "So erreichen Sie alle Bereiche Ihrer Schaftwerkzeuge, Dreh- und Stanzteile")
dieSonne-(01)-2277x1706-550x366.jpg "Fertigungsmesstechnik Shopfloor Enclosure FocusX")
