Welcome to the fascinating world of medical device regulations! If you’re a medical device manufacturer or a student stepping into this industry, you’ve probably heard about ISO 13485, the FDA’s 21 CFR Part 820, and the European Medical Device Regulation (MDR). These standards are like the guiding stars for ensuring safety, quality, and compliance. In this blog post, we’ll unravel these complex regulations in an engaging and easy-to-understand way. Buckle up as we take a deep dive into each of these critical standards!
Imagine ISO 13485 as the universal language of quality management for medical devices. It’s an internationally recognized standard that lays out the requirements for a robust quality management system (QMS). Whether you’re in Tokyo, Toronto, or Timbuktu, ISO 13485 helps ensure your medical device meets customer and regulatory expectations consistently.

1. Quality Management System (QMS): Think of this as the backbone of your operations. ISO 13485 provides a detailed roadmap for establishing a QMS that covers everything from design to delivery.
2. Management Responsibility: Top executives need to be the cheerleaders for quality. They set the tone with a quality policy, specific goals, and regular check-ins to ensure everything is on track.
3. Resource Management: Some important points of the regulations can best be explained with a comparison. Everyone has been to a restaurant before. The processes there are somewhat familiar. Think of resource management in the same way as equipping the kitchen and making sure the chefs are trained. You need the right employees, the right infrastructure, and the right environment to produce quality products.
4. Product Realization: This is the recipe for your device – planning, design, procurement, production, and monitoring. Every step ensures your product turns out just right.
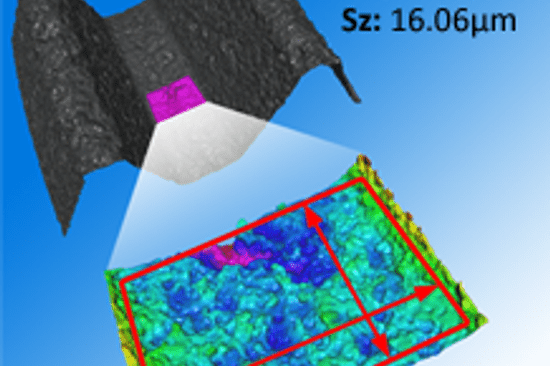
5. Measurement, Analysis, and Improvement: Constant taste-testing and tweaking. You monitor processes and products, conduct audits, manage non-conformities, and continuously improve. This is exactly where Bruker Alicona's systems are at home. It is not the taste and odour of medical products that are measured, but their size, position, shape and roughness. Read this article to find out why roughness is so important.
Global Recognition
Accepted worldwide, making it easier to enter different markets.
Customer Confidence
Shows your commitment to quality, earning trust from customers and regulators.
Continuous Improvement
Promotes a culture of ongoing enhancements.
Industry validity
As far as certification is concerned, everyone is in the same boat, there is no escape, and no one has stricter or less stringent requirements. Let's be honest: isn't it better to find such a fact cool than to get upset about the efforts?
In the U.S., medical device manufacturers must follow the FDA’s 21 CFR Part 820, also known as the Quality System Regulation (QSR). It’s like a comprehensive checklist to ensure your products are safe and effective before they hit the market.
1. Quality System Requirements: Similar to ISO 13485, this covers the entire lifecycle of a product, from initial concept to final sale.
2. Design Controls: Ensures devices are designed to meet user needs and intended use. It’s like building a house – detailed blueprints are essential.
3. Document Controls: Proper documentation for everything. Think of it as keeping meticulous records of your recipes and processes.
4. Purchasing Controls: Ensures that what you buy (ingredients, in our analogy) meets specified requirements.
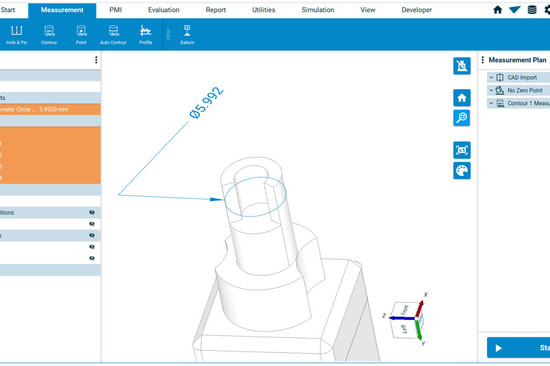
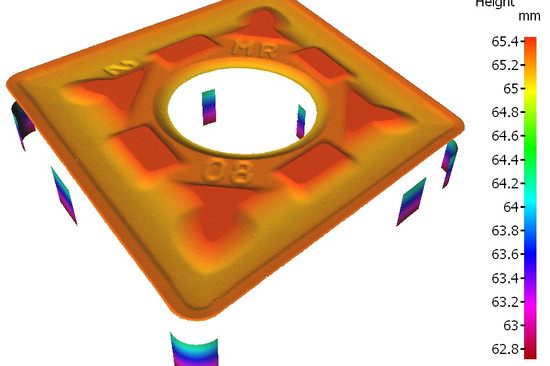
5. Production and Process Controls: Validates processes to ensure consistent quality. It’s like a chef perfecting a dish every single time. Or like the Bruker Alicona measuring system, which immediately recognizes any deviation from the drawing.
6. Corrective and Preventive Actions (CAPA): Investigates non-conformities, identifies root causes, and implements fixes. Like figuring out why a dish flopped and making sure it doesn’t happen again.
7. Records and Reports: Maintains detailed records and reports issues like adverse events. Think of it as keeping a daily journal of your culinary adventures.
In 2022, the FDA proposed harmonizing 21 CFR Part 820 with ISO 13485 by introducing the Quality Management System Regulation (QMSR). This change aims to align the FDA’s requirements more closely with the international standard, reducing the regulatory burden for global manufacturers.
ISO 13485 Alignment:
QMSR will adopt ISO 13485’s framework, streamlining compliance.
Simplified Requirements:
Reduces unique FDA requirements, focusing on ISO 13485’s comprehensive approach.
Consistency:
Easier for companies to manage global compliance.

The European Medical Device Regulation (MDR) is the EU’s stringent framework for medical devices. Replacing the old Medical Devices Directive (MDD), it’s like the EU turned up the dial on safety and performance requirements.
1. Scope and Classification: Broader scope and more devices reclassified into higher risk categories. It’s like upgrading a kitchen inspection from routine to super-scrutiny.
2. Clinical Evaluation and Investigation: Requires robust clinical data to prove safety and performance. Think of it as extensive taste tests and reviews before launching a dish.
3. Post-Market Surveillance: Continuous monitoring of devices on the market. It’s like a restaurant constantly checking customer feedback and making adjustments.
4. Unique Device Identification (UDI): A system for tracking devices. Imagine tagging each ingredient for traceability.
5. Notified Bodies: Strengthened roles for conformity assessment. It’s like having master chefs evaluate your kitchen and recipes.
6. Technical Documentation: Detailed documentation requirements. Think of it as compiling a comprehensive cookbook with every tiny detail.
Enhanced Safety
Stricter requirements ensure higher safety and performance standards.
Transparency and Traceability
Improved transparency through UDI and public access to safety data.
Global Competitiveness
Aligns with global standards, facilitating international trade.

Quality Management Focus: All three emphasize the need for a solid QMS.
Lifecycle Coverage: They cover the entire lifecycle of a device, from design to post-market activities.
Risk Management: Ensures devices are safe and effective, with robust risk management processes.
Geographical Scope: ISO 13485 is global, 21 CFR Part 820 is U.S.-specific, and the MDR applies in the EU.
Regulatory Authorities: The FDA oversees 21 CFR Part 820/QMSR, while the EU uses notified bodies for the MDR.
Specific Requirements: While ISO 13485 and 21 CFR Part 820/QMSR are closely aligned, the MDR includes unique elements like UDI and stricter clinical evaluations.
The FDA’s move to harmonize 21 CFR Part 820 with ISO 13485 through QMSR is a big win for manufacturers. It means a more straightforward regulatory landscape and less redundancy, making it easier to comply with both U.S. and international standards. The EU would also do well to harmonize the MDR with the ISO standard.
For medical device manufacturers and students, understanding ISO 13485, 21 CFR Part 820, and the MDR is crucial for navigating the complex regulatory maze. These standards and regulations ensure the safety, quality, and effectiveness of medical devices, ultimately protecting patients and enabling companies to thrive in global markets. The harmonization of FDA regulations with ISO 13485 is a significant step forward, simplifying compliance and fostering a more unified approach to quality management in the medical device industry.
So, whether you’re cooking up the next big thing in medical technology or studying to enter this dynamic field, knowing these regulations will help you ensure your devices are not just innovative but also safe, effective, and compliant. Happy innovating!
-550x366.jpeg)

die_Sonne-550x366.png "Bruker Alicona at AMB 2024 in Stuttgart")



_Nagel-550x366.jpg)


dieSonne-065-868x397-550x366.jpg)

dieSonne-120-550x366.jpg)





dieSonne-137-550x366.jpg)




-550x366.jpg)
dieSonne-(10)-2076x1706-550x366.jpg)

dieSonne-web-(102)-550x366.jpg "Rotation and tilt unit Real3DUnitX")
-550x366.jpg "knee implant measurement")
dieSonne-(23)-1706x1708-550x366.jpg "Key Regions of Turned Parts, Stamped Parts, Round Tools measured")
dieSonne-(01)-2277x1706-550x366.jpg "Production Metrology")

dieSonne-web-(21)-550x366.jpg "Turbine Blade with Cooling Holes")
